1° Passo:
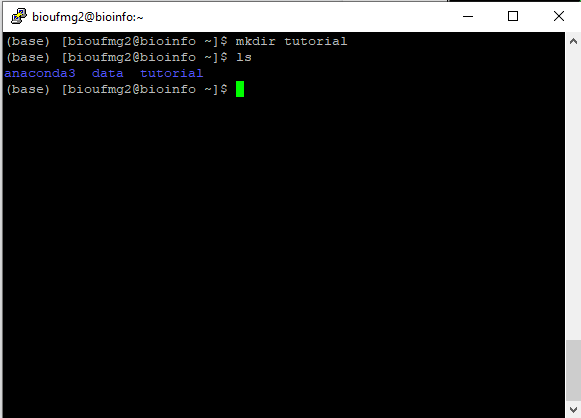
Ao entrar na bioinfo, é possível verificar que o seu home é composto de duas pastas:
Vamo criar então o nosso local de trabalho com o comando mkdir:
mkdir <seunome>
2° Passo:
Vamos copiar todos os arquivos necessários para podermos realizar esta prática. Para isso, vamos para o seu local de trabalho:
cd <seunome>
Agora vamos copiar os arquivos da pasta data:
cp -r ../data/* .
Muito importante o ponto final depois do espaço do asterisco!!
Dentro de sua pasta é possível encontrar os seguintes arquivos:
3° Passo:
Nesta aula utilizaremos dados de RNA-Seq correspondentes ao fungo Schizosaccharomyces pombe, contendo reads de 76 pares de base paired-end correspondentes a duas amostras: Sp_log (crescimento logarítmico) e Sp_plat (fase de platô). Dentro da pasta contendo essas reads, é possível encontrar os arquivos com final “left.fq” e “right.fq” no formato FASTQ formatado no estilo do sequenciador Illumina:
Para poder realizar a montagem dos transcritos, vamos utilizar todas as reads para montar um único arquivo fasta contendo todos os transcritos montados. Isto é feito para futuramente utilizarmos este arquivo para realizar a análise de genes diferenciais. Iremos rodar o Trinity com o seguinte comando:
---------------------------------------------------------------------------------------------------------------------------
Trinity --seqType fq --SS_lib_type RF --left rnaseq_data/Sp_log.left.fq.gz,rnaseq_data/Sp_plat.left.fq.gz --right rnaseq_data/Sp_log.right.fq.gz,rnaseq_data/Sp_plat.right.fq.gz --CPU 1 --max_memory 1G --output trinity_saida/
---------------------------------------------------------------------------------------------------------------------------
Detalhes:
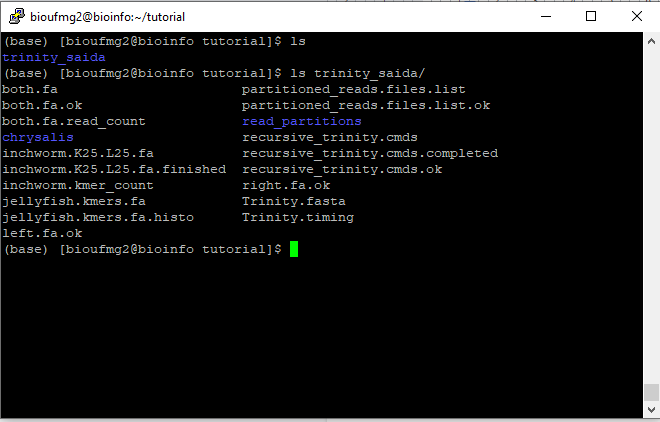
Após completar a montagem, será criado uma pasta chamada trinity_saida, tal como o seu output foi descrito. Dentro desta pasta é possível encontrar todos os dados relacionados à montagem dos transcritos.
4° Passo:
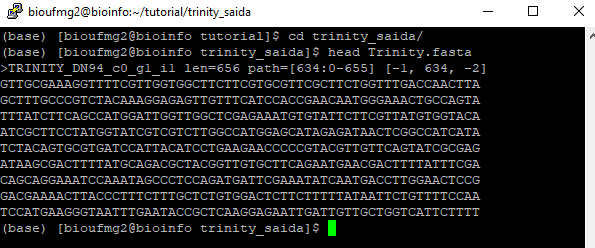
Vamos agora verificar o arquivo de montagem dos transcritos com o comando:
cd trinity_saida
less Trinity.fasta
Informações importantes:
5° Passo:
Vamos analisar agora os dados relacionados a esta montagem utilizando um script do Trinity chamado TrinityStats.pl. Para isso, vamo para a pasta do trinity_saida:
cd trinity_saida
Agora digite o comando:
TrinityStats.pl Trinity.fasta
Resultado:
################################
## Counts of transcripts, etc.
################################
Total trinity 'genes': 396
Total trinity transcripts: 400
Percent GC: 39.16
########################################
Stats based on ALL transcript contigs:
########################################
Contig N10: 2589
Contig N20: 2148
Contig N30: 1818
Contig N40: 1594
Contig N50: 1329
Median contig length: 548.5
Average contig: 839.30
Total assembled bases: 335720
#####################################################
## Stats based on ONLY LONGEST ISOFORM per 'GENE':
#####################################################
Contig N10: 2589
Contig N20: 2118
Contig N30: 1818
Contig N40: 1584
Contig N50: 1322
Median contig length: 545.5
Average contig: 830.32
Total assembled bases: 328805
Neste reporte é possível ter acesso à informações como: Número de genes encontrados, número de transcritos, a porcentagem de GC global e a média do tamanho dos contigs.
6° Passo:
Vamos agora quantificar e contar a abundância de cada transcrito e gene que foram montados utilizando o Salmon para cada condição com os seguintes comandos:
Volte agora para o seu diretório de trabalho:
cd ..
E digite os comandos:
---------------------------------------------------------------------------------------------------------------------------
/home/bioufmg2/anaconda3/pkgs/trinityrnaseq-v2.12.0/util/align_and_estimate_abundance.pl --seqType fq --left rnaseq_data/Sp_log.left.fq.gz --right rnaseq_data/Sp_log.right.fq.gz --transcripts trinity_saida/Trinity.fasta --est_method salmon --trinity_mode --prep_reference --output_dir salmon_saida/sp_log/
---------------------------------------------------------------------------------------------------------------------------
/home/bioufmg2/anaconda3/pkgs/trinityrnaseq-v2.12.0/util/align_and_estimate_abundance.pl --seqType fq --left rnaseq_data/Sp_plat.left.fq.gz --right rnaseq_data/Sp_plat.right.fq.gz --transcripts trinity_saida/Trinity.fasta --est_method salmon --trinity_mode --prep_reference --output_dir salmon_saida/sp_plat/
---------------------------------------------------------------------------------------------------------------------------
Detalhes:
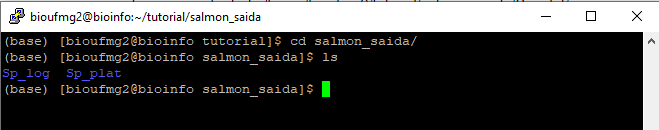
Ao final do processo, irá ser criado uma pasta chamada salmon_saida com duas pastas: Sp_log e Sp_plat, contendo todos os resultados obtidos pelo Salmon:
7° Passo:
Vamos para a pasta contendo os resultados de uma das condições para podermos analisar os arquivos de saída gerados pelo Salmon:
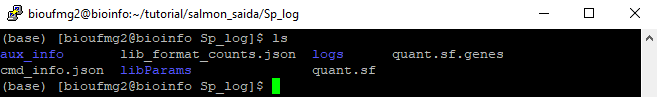
Dentro da pasta podemos encontrar 2 arquivos importantes, o quant.sf e o quant.sf.genes
Estes arquivos são separados por tabulações e possuem colunas das quais podemos identificar o nome do transcrito/gene, o tamanho, e o valores de TPM e de suas reads:
Utilizando o comando:
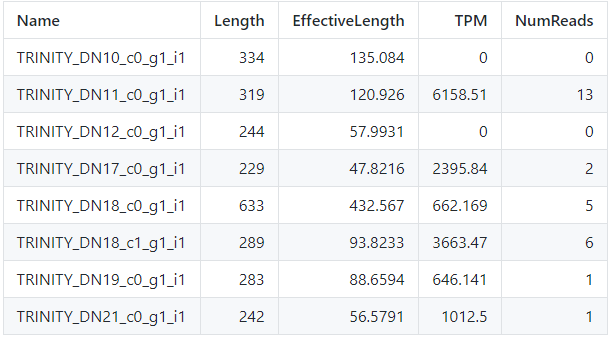
less quant.sf.genes
Obtemos o seguinte resultado (não se preocupe se o seu resultado estiver diferente da imagem, o importante são as colunas e seus valores):
Fique a vontade agora para explorar os outros arquivos.
8° Passo:
Agora vamos comparar os genes e transcritos entre as amostras utilizando um script do pacote Trinity chamado: abundance_estimates_to_matrix.pl. Primeiramente vamos para a nossa pasta de trabalho:
cd ../../
Este script irá calcular tanto a nível de genes quanto de transcritos com o seguinte comando:
--------------------------------------------------------------------------------------------------------------------------- /home/bioufmg2/anaconda3/pkgs/trinityrnaseq-v2.12.0/util/abundance_estimates_to_matrix.pl --est_method salmon --out_prefix salmon_saida/salmon salmon_saida/sp_log/quant.sf salmon_saida/sp_plat/quant.sf --gene_trans_map trinity_saida/Trinity.fasta.gene_trans_map --name_sample_by_basedir
---------------------------------------------------------------------------------------------------------------------------
Detalhes:
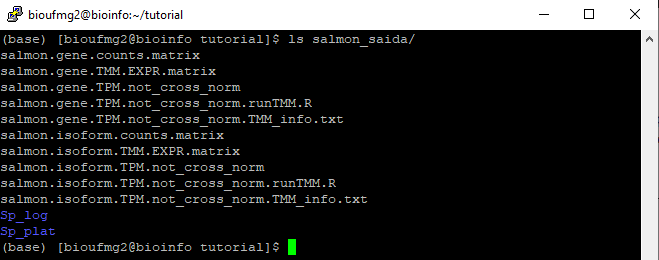
Agora com ls na pasta do salmon_saida podemos ver que apareceram novos arquivos:
Arquivos importantes:
Vamos para a salmon_saida:
cd salmon_saida
E digitar:
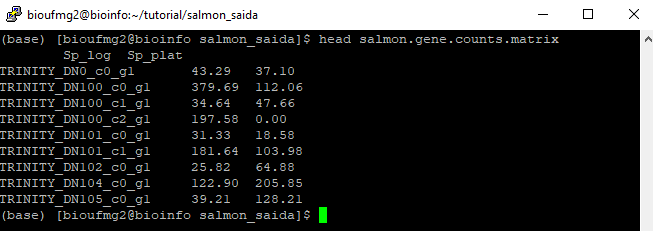
less salmon.gene.counts.matrix
Podemos observar que as colunas dos valores foram nomeadas de acordo com a pasta que elas estavam, facilitando o trabalho, nos próximos passos, para identificar qual dado é de qual amostra é condição. Chegaremos lá!
Também fique a vontade para ver os outros arquivos e perceber que todos eles seguem o mesmo padrão.
9° Passo:
Partiu agora descobrir os genes que estão diferencialmente expressos e montar uns gráficos para melhor visualização destes dados que criamos. Vamos utilizar o programa edgeR para este cálculo e outro script do Trinity chamado run_DE_analysis.pl.
Vamos agora sair da pasta do salmon_saida e vamos para a nossa pasta de trabalho:
cd ..
Certifique-se que está na sua pasta de trabalho para evitar problemas. Caso esteja perdido, utilize o comando pwd para ver o local do seu terminal.
Bora rodar:
---------------------------------------------------------------------------------------------------------------------------
/home/bioufmg2/anaconda3/pkgs/trinityrnaseq-v2.12.0/Analysis/DifferentialExpression/run_DE_analysis.pl --matrix salmon_saida/salmon.gene.counts.matrix --method edgeR --output edgeR_saida/ --samples_file samples.txt --dispersion 0.1
---------------------------------------------------------------------------------------------------------------------------
Detalhes:
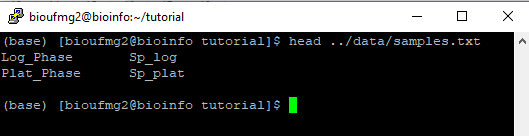
O arquivo samples.txt é muito importante nessa etapa uma vez que ele indica qual a condição e suas replicatas para o programa. Ele possui somente duas colunas, com suas linhas separadas por tabulações, e os nomes destas colunas devem estar de acordo com o nomes do passo 7, dentro do arquivo que possui o final .counts.matrix. Exemplo:
#condition #samples
condA repA1
condA repA2
condB repB1
condB repB2
10° Passo:
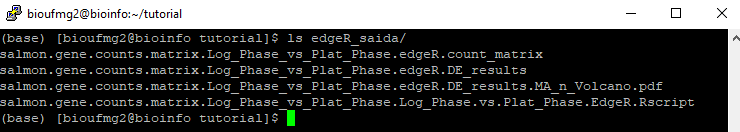
Com os cálculos do edgeR realizados, vamos agora ver os arquivos de saída. Vamos para a pasta do edgeR_saida:
cd edgeR_saida
Arquivos importantes:
Para ficar facilitar a visualização no navegador, digite o comando:
---------------------------------------------------------------------------------------------------------------------------
cp salmon.gene.counts.matrix.Log_Phase_vs_Plat_Phase.edgeR.DE_results.MA_n_Volcano.pdf ../MA_Volcano.pdf
---------------------------------------------------------------------------------------------------------------------------
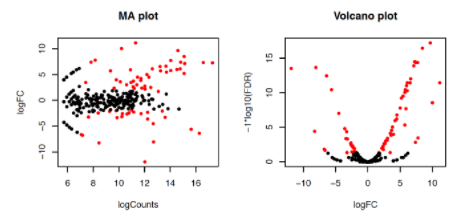
Abra agora uma nova página no seu navegador no link:
http://bioinfo.icb.ufmg.br/bioufmg/
Será necessário um login (bioufmg) e senha (carrarataxis). Ao acessar, entre na pasta look e depois a pasta com o seu nome.
Ao abrir o arquivo PDF (MA_Volcano.pdf) podemos verificar que as características com um valor de FDR < 0.5 estão coloridas de vermelho.
11° Passo:
Quantos genes estão diferencialmente expressos? Vamos descobrir com o seguinte comando (dentro da pasta edgeR_saida! pwd se estiver perdido):
---------------------------------------------------------------------------------------------------------------------------
sed '1,1d' salmon.gene.counts.matrix.Log_Phase_vs_Plat_Phase.edgeR.DE_results | awk '{ if ($7 <= 0.05) print;}' | wc -l
---------------------------------------------------------------------------------------------------------------------------
Este comando irá quantificar quantos genes possuem um valor de FDR < 0.05 (coluna 7). Sinta-se livre para brincar com este valor.
12° Passo:
Vamos agora pegar todas as sequências de todos os genes que possuem o FDR <= 0.05 utilizando o script filtrar.sh. Para isso, vamos voltar para a nossa pasta de trabalho:
cd ../
Agora só rodar:
sh filtrar.sh
Após dar um ls, é possível ver que foram criados 2 arquivos:
DE_genes.txt: contém o nome de cada gene;
DE_genes.fasta: contém o header e a sequência do gene;
13° Passo:
Agora vamos utilizar o Diamond para poder identificar a nomenclatura de cada gene para podermos conhecê-los. Para isso, certifique-se que esteja na sua pasta de trabalho. Caso esteja perdido: pwd
Para rodar o Diamond, rode o seguinte comando:
---------------------------------------------------------------------------------------------------------------------------
Diamond blastx -d ../data/uniprot_sprot.dmnd -q DE_genes.fasta -v --outfmt 6 -e 1e-10 --max-target-seqs 1 --outfmt 6 -o blastx.outfmt6 -b4
---------------------------------------------------------------------------------------------------------------------------
Detalhes:
14° Passo:
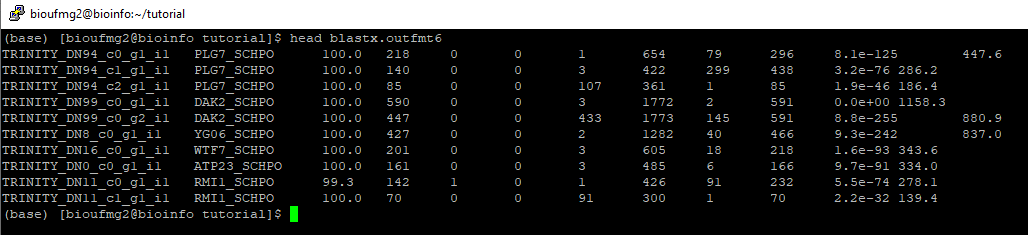
Vamos agora conhecer os nossos transcritos a partir do arquivo blastx.outfmt6. Se dermos um less neste arquivo podemos perceber que é um arquivo texto tabulado:
Nesse caso temos as seguintes colunas:
15° Passo:
Vamos agora identificar quais genes essas proteínas pertencem utilizando a plataforma online do Uniprot na aba de Get Gene Name. Primeiramente vamos printar somente a segunda coluna do nosso blast, referente aos nomes das proteínas:
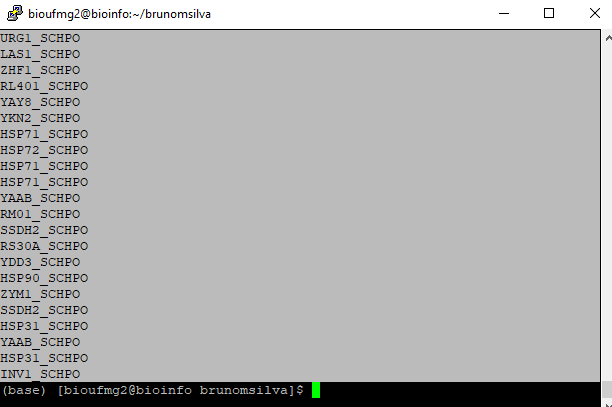
awk '{print $2}' blastx.outfmt6
Selecione todas as linhas contendo estes nomes:
Lembrando que o PuTTy copia automaticamente ao selecionar alguma coisa no terminal.
16° Passo:
Acesse agora, numa nova aba do seu navegador, o site uniprot.org.
Logo no cabeçalho do site vamos clicar em Retrieve/ID mapping:
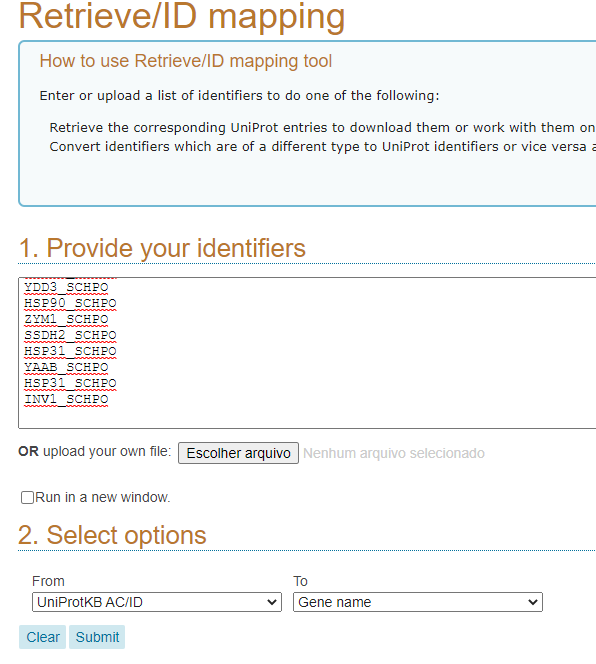
Cole o texto que você copiou do PuTTY (ctrl + v) na região do Provide your identifiers; selecione, na parte de Select Options, To Gene Name. Por fim, clique em Submit.
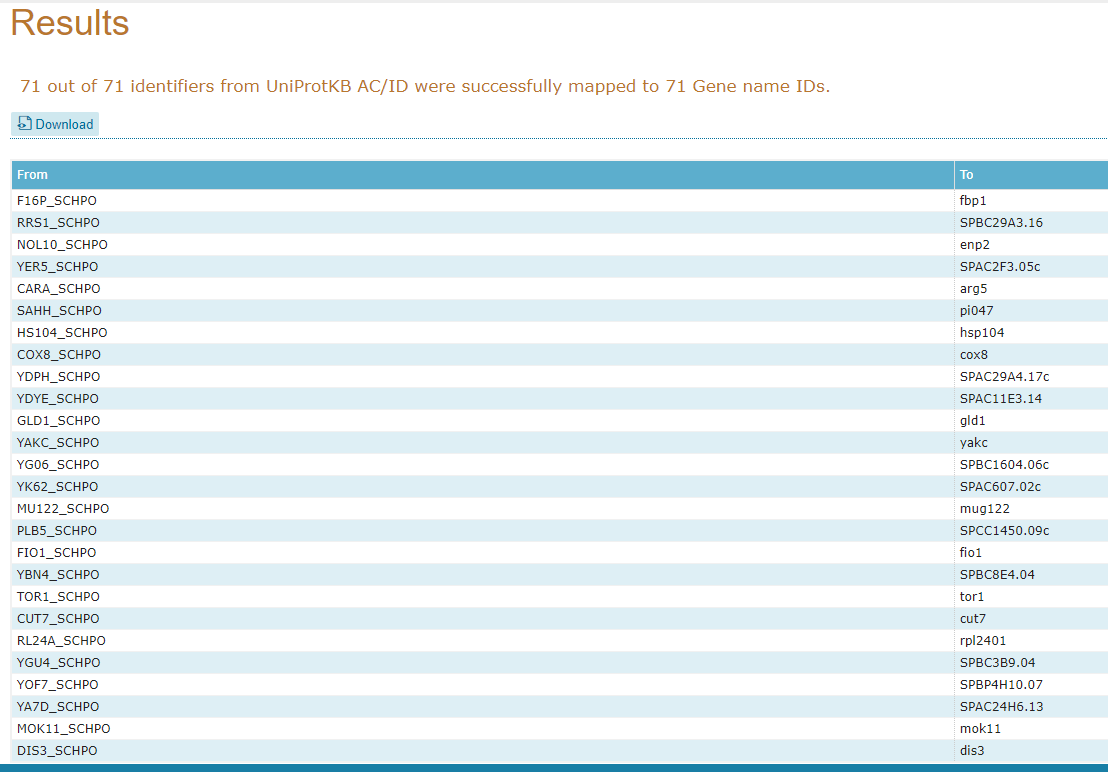
Irá abrir então uma página contendo todos os gene names dos seus identificadores:
17° Passo:
Retorne à página anterior do seu browser para fazermos uma nova busca utilizando os mesmos identificadores. Desta vez, vamos selecionar To KEGG dentro do grupo de Genome:
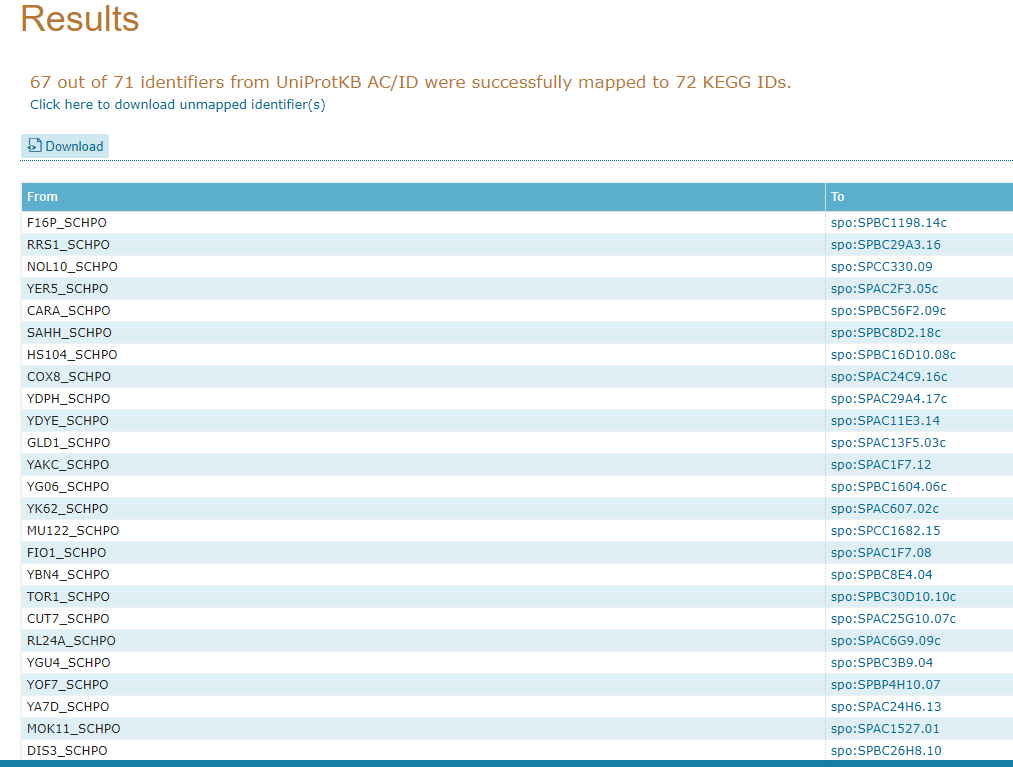
Agora irá abrir uma página contendo os identificadores KEGG que dão acesso diretamente a esse banco de dados:
18° Passo:
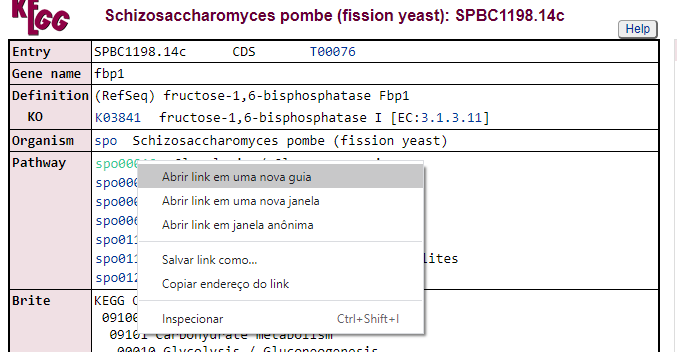
Vamos explorar alguma dessas vias. Clique na primeira opção de KEGG com o botão direito e vá em Abrir em uma nova guia:
Na aba que irá abrir, será do banco de dados do KEGG:
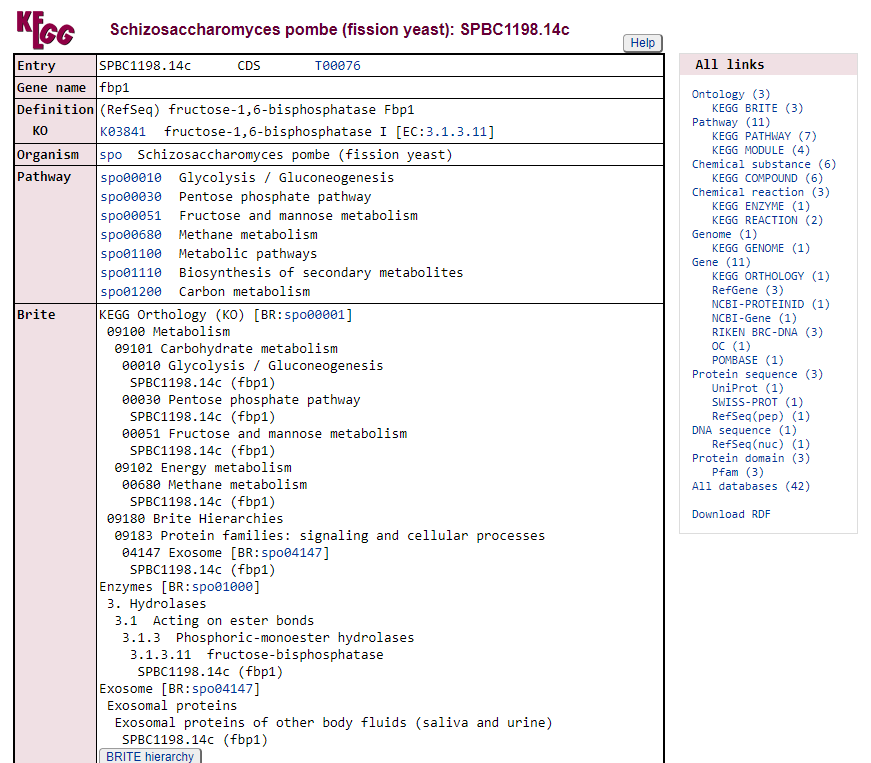
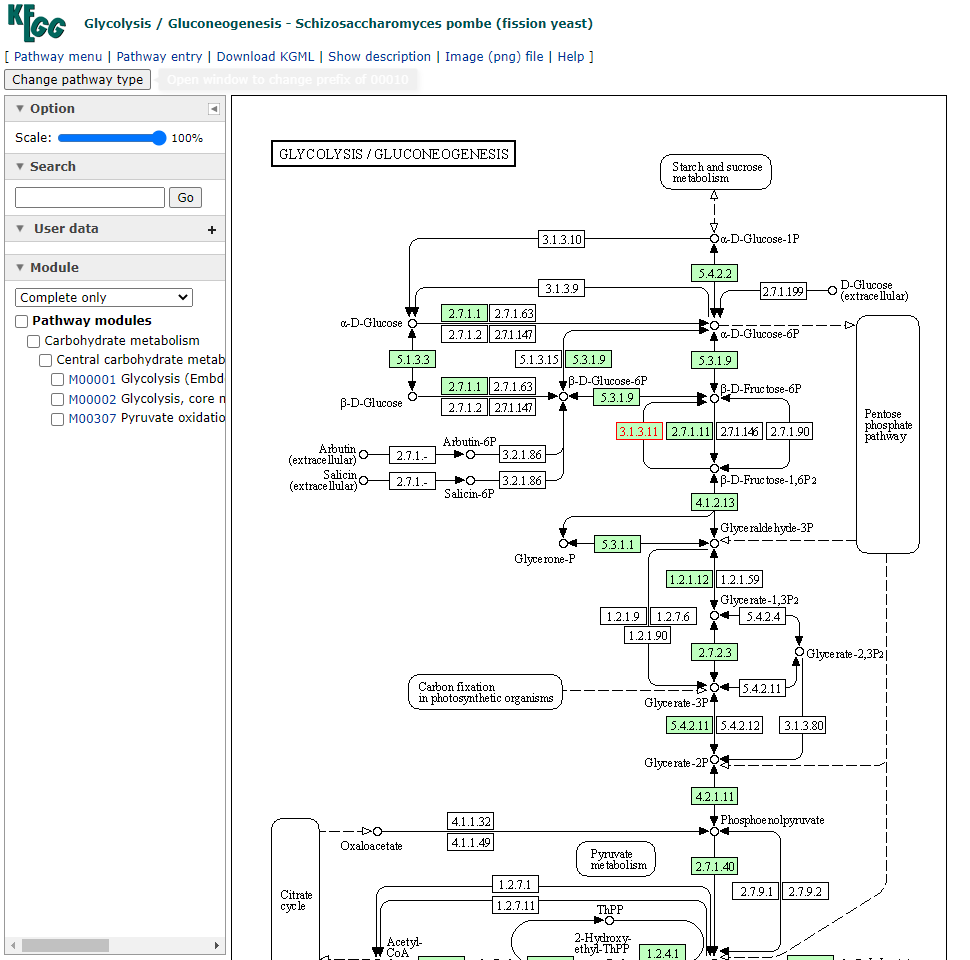
A partir daqui nós podemos visualizar todas as vias metabólicas que este gene está envolvido, como por exemplo, a spo00010 Glycolysis/Gluconeogenesis. Vamos fazer o mesmo procedimento poder abrir este link em uma nova aba do navegador:
Nesta nova aba aberta agora podemos explorar em qual região da via este gene está com sua expressão diferenciada. Neste caso, está em vermelho o número 3.1.3.11 dentro da parte de Pentose phophate pathway.
Agora você pode explorar todas as vias e genes que você identificou como diferencialmente expressos. Boa sorte.
PASSOS ADICIONAIS OPTATIVOS:
19° Passo:
Iremos agora extrair esses genes e gerar os famosos gráficos de heatmap. Para isso utilizaremos o analyze_diff_expr.pl do Trinity, mas precisamos estar dentro da pasta edgeR_saida:
---------------------------------------------------------------------------------------------------------------------------
/home/bioufmg2/anaconda3/opt/trinity-2.1.1/Analysis/DifferentialExpression/analyze_diff_expr.pl -P 1e-3 -C 2 --matrix ../salmon_saida/salmon.gene.TMM.EXPR.matrix --samples ../samples.txt
---------------------------------------------------------------------------------------------------------------------------
Detalhes:
Dando um ls nós vemos vários arquivos novos que apareceram:
Arquivos importantes:
Vamos agora brincar com estes arquivos.
20° Passo:
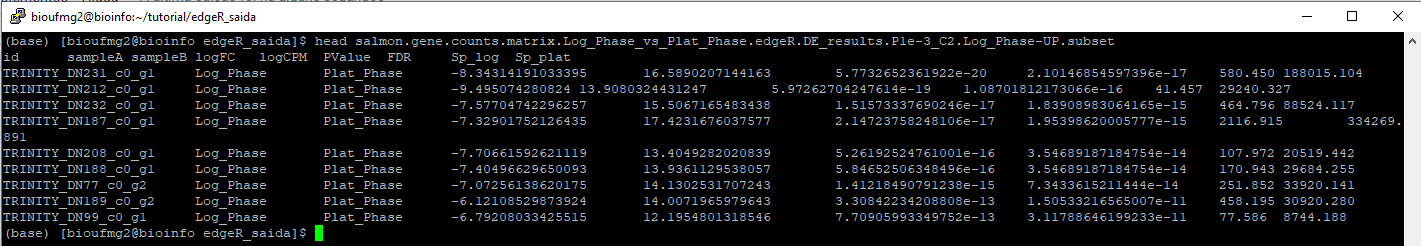
Vamos conhecer os genes super regulados em uma amostra com um:
---------------------------------------------------------------------------------------------------------------------------
less salmon.gene.counts.matrix.Log_Phase_vs_Plat_Phase.edgeR.DE_results.P1e-3_C2.Log_Phase-UP.subset
---------------------------------------------------------------------------------------------------------------------------
Lembre-se de estar dentro da pasta edgeR_saida. Podemos perceber que este arquivo possui a separação em suas linhas por tabulações, facilitando a manipulação destes arquivos. Fique a vontade para conhecer os genes da outra amostra!
21° Passo:
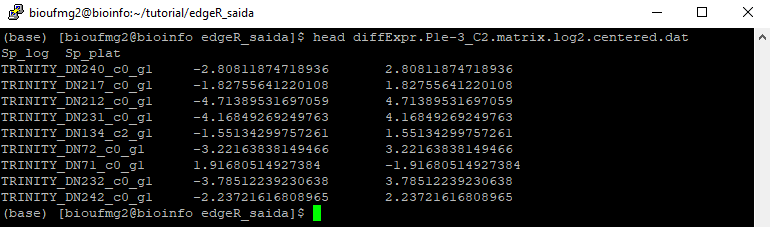
Com um less diffExpr.P1e-3_C2.matrix.log2.centered.dat nós podemos perceber que este arquivo também é tabulado:
22° Passo:
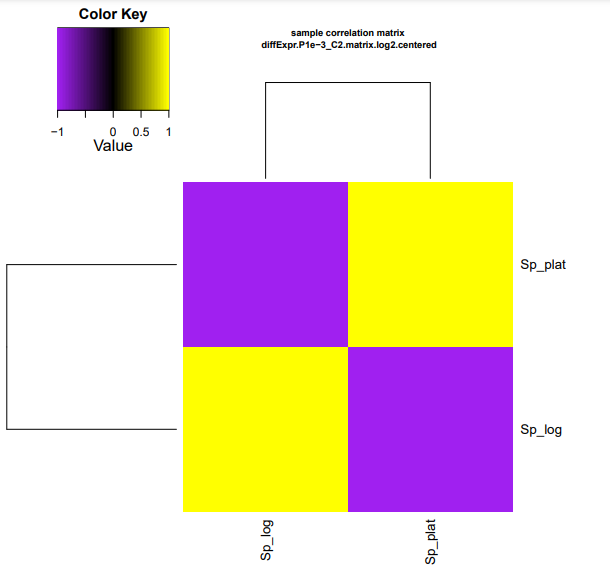
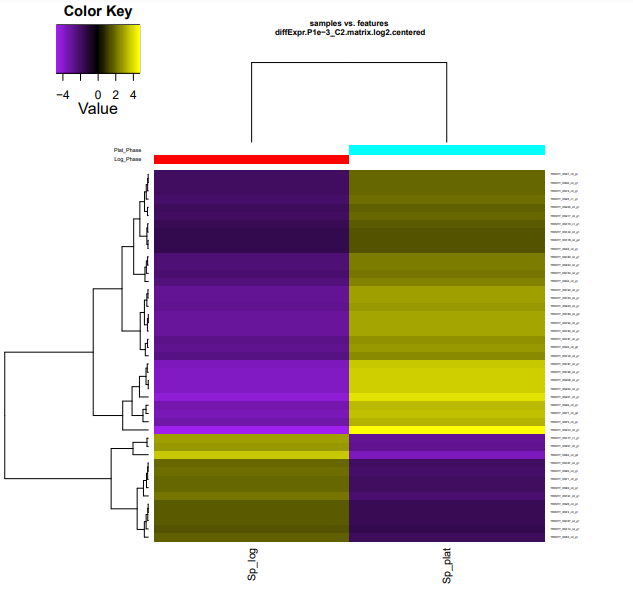
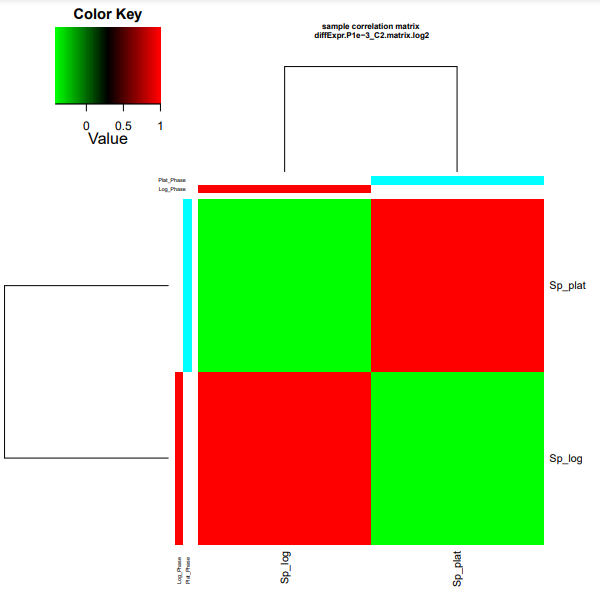
Os resultados dos seus heatmaps gerados devem se parecer com os mostrados abaixo.
Para facilitar visualização no navegador, digite os comandos dentro da pasta edgeR_saida:
---------------------------------------------------------------------------------------------------------------------------
cp diffExpr.P1e-3_C2.matrix.log2.centered.genes_vs_samples_heatmap.pdf ../heatmap_cluster.pdf
cp diffExpr.P1e-3_C2.matrix.log2.sample_cor_matrix.pdf ../heatmap_cor_matrix.pdf
cp diffExpr.P1e-3_C2.matrix.log2.centered.sample_cor_matrix.pdf ../heatmap_centred_matrix.pdf
---------------------------------------------------------------------------------------------------------------------------
23° Passo:
Agora vamos terminar as análises de genes diferenciados e fazer uma clusterização dos mesmos para melhor visualização de grupos que com um padrão similar nas amostras!
Vamos dar o seguinte comando, ainda dentro da pasta edgeR_saida:
---------------------------------------------------------------------------------------------------------------------------
define_clusters_by_cutting_tree.pl -R diffExpr.P1e-3_C2.matrix.RData --Ktree 3
---------------------------------------------------------------------------------------------------------------------------
Detalhes:
Foi criado agora uma nova pasta, vamos para ela.
24° Passo:
Dentro da pasta edgeR_saida vamos dar um:
cd diffExpr.P1e-3_C2.matrix.RData.clusters_fixed_Ktree_3/
Lembrando que o nome da pasta depende dos valores de p-valor, FC e Ktree utilizados nos passos do script analyze_diff_expr.pl e define_clusters_by_cutting_tree.pl!
25° Passo:
Observando os arquivos desta pasta podemos verificar que foi adicionado um novo arquivo pdf e outros de texto.
Para facilitar, vamos dar o seguinte comando, dentro da pasta diffExpr.P1e-3_C2.matrix.RData.clusters_fixed_Ktree_3/:
---------------------------------------------------------------------------------------------------------------------------
cp my_cluster_plots.pdf ../../my_cluster_plots.pdf
---------------------------------------------------------------------------------------------------------------------------
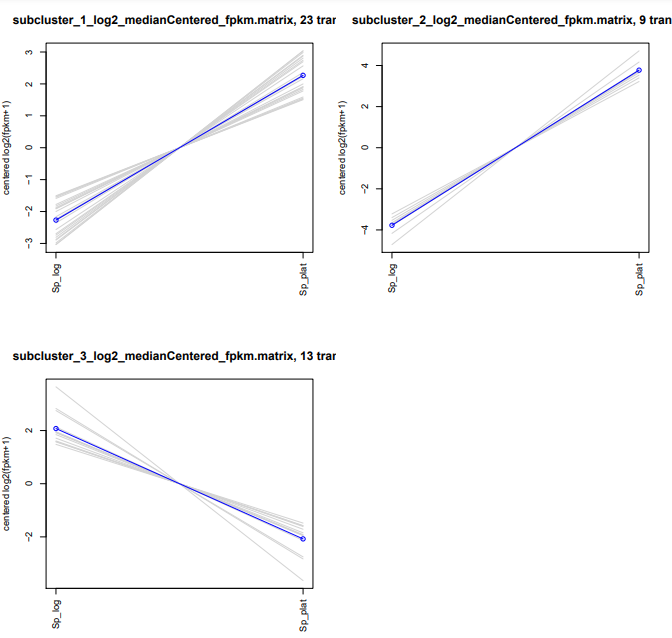
O seu PDF deve se parecer com algo assim:
26° Passo:
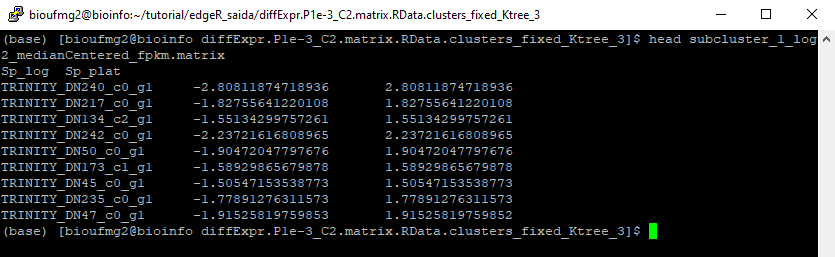
Para poder saber exatamente quem são esses grupos, basta somente verificar cada arquivo que foi gerado a partir do algoritmo de clusterização com o comando que preferir!
Para cada cluster é gerado este tipo de arquivo tabulado. Caso tenha curiosidade, você pode explorar os outros sub-clusters criados.